BRGenomics includes useful utilities for spike-in normalization.
A typical approach is to add the spike-in (either exogenous cells or synthetic oligonucleotides) before library preparation, and to subsequently map to a combined genome containing both the target organisms chromosomes (to map the experimental reads) as well as sequences/chromosomes for the spike-in.
This so-called “competitive alignment” results in the creation of BAM files containing a mix of chromosomes, for which it should be straightforward to identify the spike-in chromosomes.
Counting Spike-in Reads
For this section, we’ll use a list of 4 dummy datasets containing normal, as well as spike-in chromosomes. Consider the first 2 datasets:
grl[1:2]## $gr1_rep1
## GRanges object with 4 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 1
## [2] chr2 2 + | 1
## [3] spikechr1 3 + | 1
## [4] spikechr2 4 + | 1
## -------
## seqinfo: 4 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep1
## GRanges object with 4 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 2
## [2] chr2 2 + | 2
## [3] spikechr1 3 + | 1
## [4] spikechr2 4 + | 1
## -------
## seqinfo: 4 sequences from an unspecified genome; no seqlengthsWe can identify the spike-in chromosomes either by full names, or by a regular expression that matches the spike-in chromosomes. In this case, we named our spike-in chromosomes to contain the string “spike” which makes them easy to identify.
To count the reads for each dataset:
getSpikeInCounts(grl, si_pattern = "spike", ncores = 1)## sample total_reads exp_reads spike_reads
## 1 gr1_rep1 4 2 2
## 2 gr2_rep1 6 4 2
## 3 gr1_rep2 5 2 3
## 4 gr2_rep2 12 8 4Filtering Spike-in Reads
We can also remove the spike-in reads from our data:
removeSpikeInReads(grl[1:2], si_pattern = "spike", ncores = 1)## $gr1_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 1
## [2] chr2 2 + | 1
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 2
## [2] chr2 2 + | 2
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsAnd if we wanted to isolate the spike-in reads, there is an analogous getSpikeInReads() function.
The Spike-in Normalization Factor
There are several methods by which to generate spike-in normalization factors, but we advocate for a particular method, which generates units we call Spike-in normalized Reads Per Million mapped reads in the negative Control (SRPMC). The SRPMC normalization factor for a given sample \(i\) is defined as such:
\[SRPMC:\ NF_i = \frac{\sum reads_{spikein, control}}{\sum reads_{spikein, i}} \cdot \frac{10^6}{\sum reads_{experimental, control}}\]
This expression effectively calculates Reads Per Million (RPM) normalization for the negative control, and all other samples \(i\) are scaled into equivalent units according to the ratio of their spike-in reads. We provide a more explicit derivation below.
Derivation of SRPMC
The fundamental concept of spike-in normalization is that the ratio of experimental reads to spike-in reads can be used to correct for global changes in starting material. Let’s call this ratio Reads Per Spike-in read (RPS):
\[RPS = \frac{\sum reads_{experimental}}{\sum reads_{spikein}}\]
In isolation, this number only reflects the relative amounts of spike-in material recovered and mapped. The meaningful information about changes in material can only arise from making direct comparisons between samples. For any sample, \(i\), we can calculate the global change in signal as a proportion of the material recovered from a negative control:
\[RelativeSignal_i = \frac{RPS_i}{RPS_{control}}\]
The usual purpose of spike-in normalization is to measure a biological difference in total material (e.g. RNA) between samples, and the above ratio is a direct measurement of this.
To generate normalization factors, we use the above ratio to adjust RPM (Reads Per Million mapped reads) normalization factors, which we define below for clarity:
\[RPM:\ NF_i = \frac{1}{\frac{\sum{reads_i}}{10^6}} = \frac{10^6}{\sum{reads_i}}\]
(Unless indicated, \(reads\) refers to non-spike-in reads).
RPM normalization (i.e. read depth normalization) is the simplest and likely most familiar form of normalization. For a basal (unperturbed) negative control, RPM should produce the most portable metric of signal, given that we intend for the negative control to demonstrate typical physiology, and we hope that this state is reproducible. We therefore want to have our normalized signal in unit terms of RPM in the negative control.
To accomplish this, we multiply the ratio of spike-in normalized reads between the sample \(i\) and the negative control to the RPM normalization factor for sample \(i\). This converts readcounts into units we summarize as Spike-in normalized Reads Per Million mapped reads in the negative Control (SRPMC):
\[SRPMC:\ NF_i = \frac{RPS_i}{RPS_{control}} \cdot \frac{10^6}{\sum{reads_i}}\]
Again, SRPMC results in the negative control being RPM (sequencing depth) normalized, while all other samples are in equivalent, directly comparable units. And we’ve effectively determined the relative scaling of those samples based on the ratios of spike-in reads.
This becomes more apparent if we substitute the \(RPS\) variables above. \(\sum{reads_i}\) cancels, and simplifying the fraction produces the original formula:
\[SRPMC:\ NF_i = \frac{\sum reads_{spikein, control}}{\sum reads_{spikein, i}} \cdot \frac{10^6}{\sum reads_{experimental, control}}\]
Calculating Normalization Factors
We can calculate SRPMC normalization factors for each sample using the getSpikeInNFs() function, using the same syntax we used to count the spike-in reads.
However, we have to also identify the negative control, which is the sample that will have the “reference” (RPM) normalization. We do this either using a regular expression (ctrl_pattern argument) or by supplying the name(s) of the negative controls (the ctrl_names argument).
The default method is “SRPMC”, but there are other options, as well.
getSpikeInNFs(grl, si_pattern = "spike", ctrl_pattern = "gr1", ncores = 1)## [1] 500000 500000 500000 375000(The NFs are high because our dummy data contain only a few reads).
By default, normalization factors utilize batch normalization, such that in any replicate (identified by the characters following “rep” in the sample names), the negative control is RPM normalized, and the other conditions are normalized to the within-replicate negative control (see the documentation for further details).
Currently, batch normalization requires the sample names end with strings matching the format "_rep1", "_rep2", etc. If sample names do not conform to this pattern, you can rename them by using the sample_names argument.
Normalizing Data
We can also use the spikeInNormGRanges() function to simultaneously find the spike-in reads, calculate the spike-in normalization factors, filter out spike-in reads, and normalize the readcounts:
spikeInNormGRanges(grl, si_pattern = "spike", ctrl_pattern = "gr1", ncores = 1)## $gr1_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 5e+05
## [2] chr2 2 + | 5e+05
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 1e+06
## [2] chr2 2 + | 1e+06
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr1_rep2
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 5e+05
## [2] chr2 2 + | 5e+05
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep2
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 1500000
## [2] chr2 2 + | 1500000
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsNormalization by Sub-sampling
Rationale
When viewing genomics data in a genome browser (or otherwise plotting signal for a single gene), the sparsity of basepair resolution data can challenge our visual perception.
Consider two datasets from identical samples, but where one is sequenced to a higher depth. The two datasets can be normalized such that the signal counts are equivalent, but the dataset with higher sequencing depth will have also uncovered additional sites. When plotted in a genome browser, the total signal within a region may be the same, but the more highly sequenced dataset will cover more positions but have lower peaks, while the less sequenced dataset will look sparse and spikey in comparison.
Below, we compare PRO-seq data derived from the same dataset over the same gene. In one case, we randomly sample half of the reads over that gene, while in another, we divide all the readcounts by 2.
# choose a single gene
gene_i <- txs_dm6_chr4[185]
reads.gene_i <- subsetByOverlaps(PROseq, gene_i)
# sample half the raw reads
set.seed(11)
sreads.gene_i <- subsampleGRanges(reads.gene_i, prop = 0.5, ncores = 1)
# downscale raw reads by a factor of 2
score(reads.gene_i) <- 0.5 * score(reads.gene_i)
plot(x = 1:width(gene_i),
y = getCountsByPositions(sreads.gene_i, gene_i),
type = "h", ylim = c(0, 20),
main = "PRO-seq (down-sampled)",
xlab = "Distance from TSS", ylab = "Down-sampled PRO-seq Reads")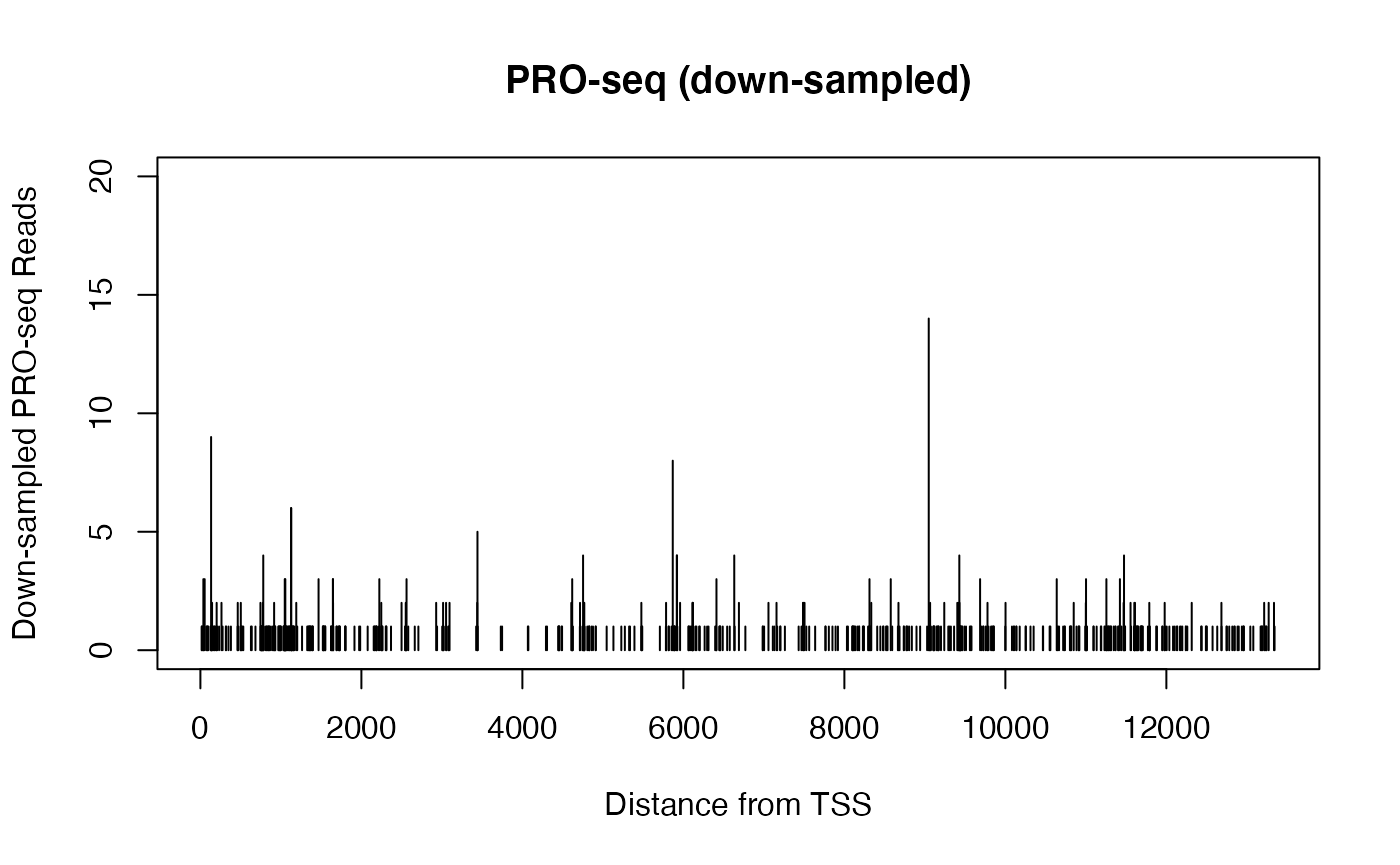
plot(x = 1:width(gene_i),
y = getCountsByPositions(reads.gene_i, gene_i),
type = "h", ylim = c(0, 20),
main = "PRO-seq (down-scaled)",
xlab = "Distance from TSS", ylab = "Down-scaled PRO-seq Reads")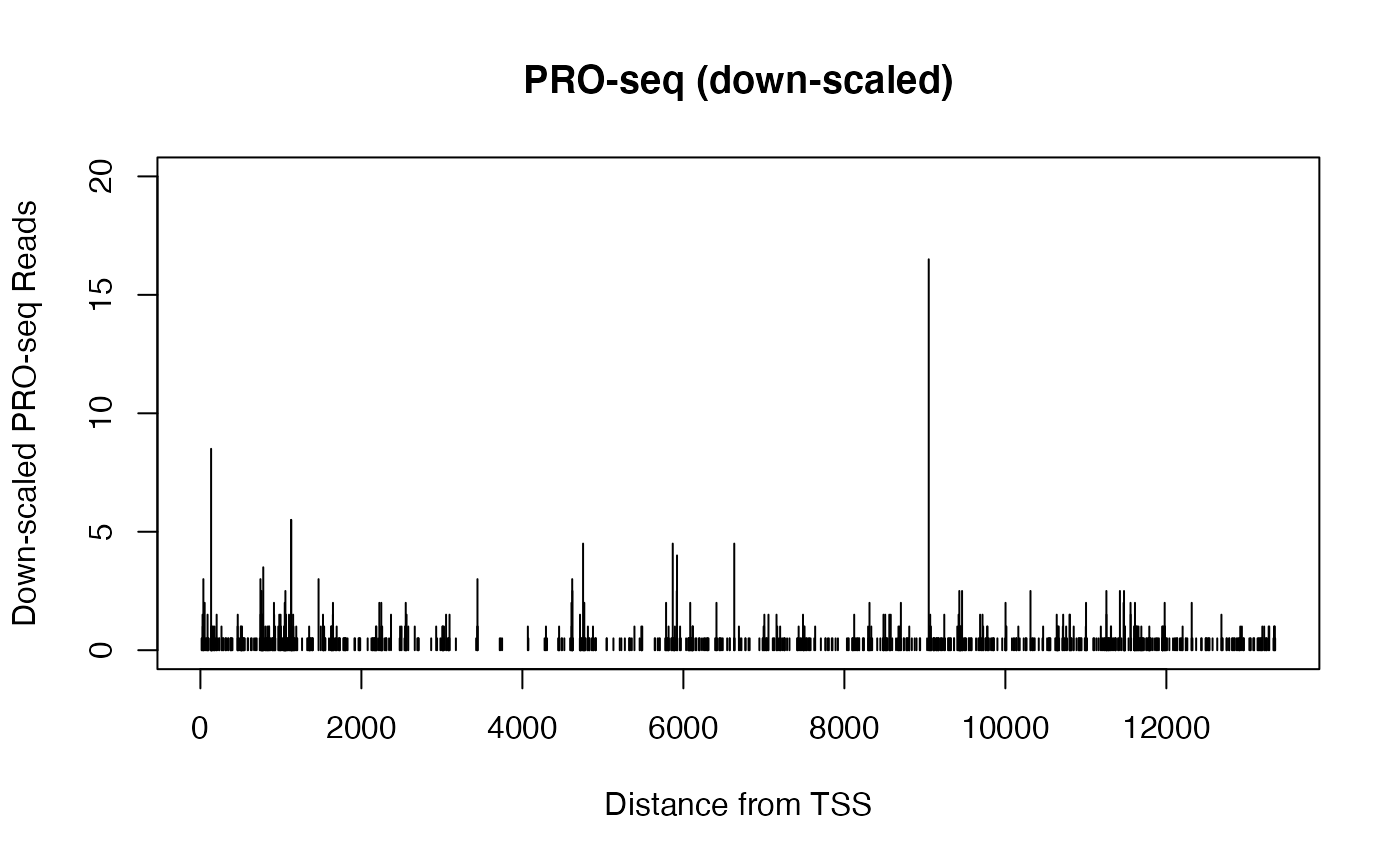
The two plots above come from the same data, and contain the same quantity of signal, but their profiles are notably distinct. Particularly when plotting many samples over large regions within a genome browser, differences caused by sequencing depth can be misleading. It can be challenging to estimate differences if some datasets are “tall and spikey” while others are “short and smooth”.
Absent global changes in signal, the above scenario can be resolved beforehand by equal sequencing depths, or by down-sampling to match readcounts.
However, matching raw readcounts is not a solution when significant biological changes in total signal should be accounted for.
For instance, consider an example in which there is a true, two-fold biological difference in transcription between two samples. If we could avoid all technical artifacts and measure the transcription directly in each individual cell, we would expect to uncover half the number of transcribing complexes in the lower condition. Having equivalent sequencing depth across those two conditions is effectively a technical artifact, and down-scaling the signal by multiplication can cause the visual challenges observed above.
Sub-sampling for Normalization
To address the above concerns, we’ve included a function subsampleBySpikeIn() to randomly sample reads to match the normalized signal proportions between datasets.
Internally, the function uses the getSpikeInNFs() function, but instead of SRPMC normalization, using the option method = "SNR", which calculates normalization factors that downscale each dataset to match the dataset with the least spike-in reads. From this, the number of “desired reads” is established for each dataset, and subsequently that number of reads is randomly sampled.
removeSpikeInReads(grl, si_pattern = "spike", ncores = 1)## $gr1_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 1
## [2] chr2 2 + | 1
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 2
## [2] chr2 2 + | 2
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr1_rep2
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 1
## [2] chr2 2 + | 1
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep2
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <numeric>
## [1] chr1 1 + | 4
## [2] chr2 2 + | 4
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
getSpikeInNFs(grl, si_pattern = "spike", method = "SNR", batch_norm = FALSE,
ncores = 1)## [1] 1.0000000 1.0000000 0.6666667 0.5000000
subsampleBySpikeIn(grl, si_pattern = "spike", batch_norm = FALSE, ncores = 1)## $gr1_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <integer>
## [1] chr1 1 + | 1
## [2] chr2 2 + | 1
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep1
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <integer>
## [1] chr1 1 + | 2
## [2] chr2 2 + | 2
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr1_rep2
## GRanges object with 1 range and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <integer>
## [1] chr1 1 + | 1
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengths
##
## $gr2_rep2
## GRanges object with 2 ranges and 1 metadata column:
## seqnames ranges strand | score
## <Rle> <IRanges> <Rle> | <integer>
## [1] chr1 1 + | 1
## [2] chr2 2 + | 3
## -------
## seqinfo: 2 sequences from an unspecified genome; no seqlengthsNormalization by sub-sampling sacrifices information to reduce biases across datasets.